cro-西典科技-cro产品引进
临床III期
Ⅲ期临床试验
在Ⅰ,Ⅱ期临床研究的基础上,将试验*用于更大范围的病人志愿者身上,进行扩大的多中心临床试验,进一步评价*的有效性和耐受性(或安全性),称之为Ⅲ期临床试验。Ⅲ期临床试验可以说是治l疗作用的确证阶段,也是为药品注册申请获得批准提供依据的关键阶段,该期试验一般为具有足够样本量的随机化盲法对照试验。临床试验将对试验*和安慰剂(不含活性物质)或已上市药品的有关参数进行比较。试验结果应当具有可重复性。可以说,该阶段是临床研究项目的****繁忙和任务****集中的部分。除了对成年病人研究外,还要特别研究*对老年病人,cro制药研发,有时还要包括儿童的安全性。一般来讲,老年病人和危重病人所要求的剂量要低一些,因为他们的身体不能有产地清除*,使得他们对不良反应的耐受性更差,所以应当进行特别的研究来确定剂量。而儿童人群具有突变敏*、迟发毒性和不同的*代谢动力学性质等特点,因此在决定*应用于儿童人群时,权衡疗l效和*不良反应应当是一个需要特别关注的问题。在国外,儿童参加的临床试验一般放在*试验的Ⅲ期临床后才开始。如果一种疾病主要发生在儿童,并且很严重又没有其他治l疗方法,美*品与药品管l理局允许Ⅰ期临床试验真接从儿童开始,即在不存在*数据参照的情况下,允许从儿童开始药理评价。我国对此尚无明确规定。

参比制剂的选择和确定
1.药品生产企业对拟进行一致性评价的品种,参照《普通口服固体制剂参比制剂选择和确定指导原则》(食品药品监管总局公告2016年第61号)要求选择参比制剂。2.药品生产企业按照《仿l制药质量和疗l效一致性评价参比制剂备案与推荐程序》(食品药品监管总局公告2016年第99号),将选择的参比制剂向食品药品监管总局仿l制药质量一致性评价办公室(以下简称一致性评价办公室)备案。行业协会可向一致性评价办公室推荐参比制剂,cro,原研药品生产企业、国际公认的同种*生产企业可向一致性评价办公室申报参比制剂。一致性评价办公室主动对参比制剂的备案、推荐和申报信息向社会公开。食品药品监管总局及时公布推荐和确定的参比制剂信息,药品生产企业原则上应选择公布的参比制剂开展一致性评价。3.企业找不到且无法确定参比制剂的,由药品生产企业开展临床有效性试验。

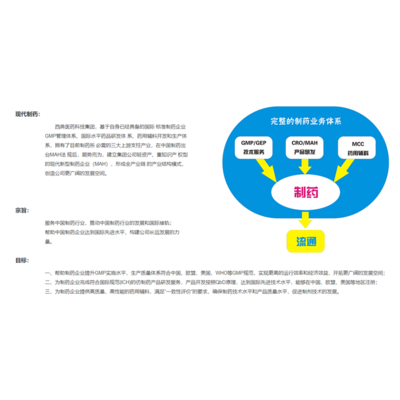
仿l制药一致性评价是指对已经批准上市的仿l制药,按与原研药品质量和疗l效一致的原则,分期分批进行质量一致性评价,cro产品引进,就是仿l制药需在质量与*上达到与原研药一致的水平。对已经批准上市的仿l制药进行一致性评价,这是补历史的课。因为过去批准上市的药品没有与原研药一致性评价的强制性要求,有些药品在疗ll效上与原研药存在一些差距。历史l上,美国、日本等*也都经历了同样的过程,日本用了十几年的时间推进仿l制药一致性评价工作。开展仿l制药一致性评价,可以使仿l制药在质量和疗ll效上与原研药一致 ,在临床上可替代原研药,这不仅可以节约医l疗费用,同时也可提升我国的仿l制药质量和制药行业的整体发展水平,保证公众用药安全有效。
cro-cro产品技术转移-西典科技(****商家)由东莞西典医药科技有限公司提供。东莞西典医药科技有限公司(www.ttgwp*)实力雄厚,信誉可靠,在广东 东莞 的其它等行业积累了大批忠诚的客户。公司精益求精的工作态度和不断的完善*理念将*西典科技和您携手步入*,共创美好未来!